|
|
Clinical Pharmacology of SSRI's
7 - Why Are CYP Enzymes Important When Considering SSRIs? |
|
|
|
|
|
Cytochrome P450 (CYP) enzymes may be termed an "overnight
discovery" a billion years in the making (Table
7.1). Only recently have we begun to understand the important
role these enzymes play in determining a patient's response
to pharmacotherapy. The inhibition of specific CYP enzymes
is also the major distinguishing characteristic among SSRIs.
Such inhibition produces the potential for specific pharmacokinetic
drug interactions between specific SSRIs and concomitantly
administered drugs dependent on specific CYP enzymes for their
elimination. This section will provide the background information
of how our understanding of these enzymes has evolved and
their significance relative to the optimal
care of patients.
| TABLE 7.1 Unintended Targets
of Some SSRIs: CYP Enzymes |
- Evolved over 1 billion years ago
- Expressed in multiple tissues
- Highest concentration in hepatocytes
- Located in:
- Mitochondria: steroidogenic CYP enzymes
- Endoplasmic reticulum: xenobiotic CYP enzymes
- Mediate primarily oxidative metabolism
|
| References: 105,
115, 116,
183-185, 187 |
Our knowledge of oxidative metabolism in the body and the
role played by CYP enzymes has been relatively short, but
has rapidly expanded, particularly over the last
10 years (Table 7.2). It was only about
100 years ago that drug metabolism was generally accepted
to occur in the body. The pigments we now recognize to be
multiple, different enzymes were identified approximately
50 years ago and named cytochrome P450 (CYP) due to their
ability to absorb light at a frequency of 450 nm. The first
gene to code for a specific CYP enzyme was isolated approximately
10 years ago. Due to the advances made possible by molecular
biology, we can now study the effects of specific CYP enzymes
on specific drugs and vice versa. We are currently "backfilling"
our knowledge concerning which CYP enzymes are responsible
for the metabolism of drugs.
Although our knowledge is relatively recent, the ancestral
CYP enzyme from which all of the current ones evolved came
into existence over 1 billion years ago, which underscores
the biological importance of these enzymes to organisms, including
man.106,107
They are heme-containing monoxygensase enzymes responsible
for much of the oxidative metabolism occurring in the body.105
Once the genes that code for specific CYP enzymes were isolated,
they could be used to produce the enzymes in purified form
- their amino acid sequence could then be determined. This
has now been accomplished for all of the human CYP enzymes.
| TABLE 7.2
History of Our Knowledge of Biotransforming Enzymes |
| Knowledge |
Time |
Concept of entgiftung
(detoxification)
was appreciated
Correlation of some drug effects with
in vitro metabolizing enzyme activities
Induction of such enzymes possible
Membrane bound cytochrome-CO complex
discovered, named "cytochrome
P450,"
and biochemically characterized
Three hundred substances identified to
induce their metabolism or that
of
foreign and endogenous substances
Role of mRNA and protein synthesis
identified in induction process
and
purification of multiple P450
proteins
accomplished, making polyclonal
and
monoclonal antibody studies possible
Identification of specific P450 genes using
DNA cloning techniques |
End of the
19th century
1920s-40s
1940s-50s
1950s-60s
Mid-1960s
1970s
Mid-1980s
to present |
| Reference:
218 |
|
 |
| TABLE 7.3
Human CYP Enzymes as Classified by Family, Subfamily
and Gene* |
1A1
1A2 |
2A6
2A7
2B6
2C8
2C9
2C18
2C19
2D6
2E1
2F1 |
3A3/4
3A5
3A7 |
4A9
4B1
4F2
4F3 |
7 |
11A1
11B1
11B2 |
17 |
19 |
21A2 |
27 |
| * Key to classification:
1) The first Arabic numeral represents the family,
2) the alphabetic letter represents the subfamily,
and 3) the second Arabic numeral represents the
individual gene within the subfamily. |
| From reference:
231 |
|
|
|
| TABLE 7.4
Two General Classes of CYP Enzymes |
Families 1, 2, 3, 4
Xenobiotic CYP Enzymes
Evolved from steroidogenic CYP enzymes during
the period of plant-animal differentiation
Co-evolved with transferase enzymes
Degrade dietary toxins produced by plants
Biotransforms drugs to permit elimination |
Families 7, 11, 17,
19, 21, 27
Steroidogenic CYP Enzymes
For membrane integrity of single cell organisms
For hormonal mediators of the development of differentiated
organisms
With rigid substrate and product specificity |
| Data from references: 105,
115, 116,
183-185, 187 |
|
|
|
| TABLE 7.5 What Are The
Functions of CYP Enzymes? |
Steroidogenic:
conversion of consumed substances into biologically
needed constituents:
Steroids
Bile acids
Cholesterol
Prostaglandin biosynthesis
Xenobiotic: detoxification of consumed
substances:
Toxins
Carcinogens
Drugs
Mutagens |
| Data from references:
105, 115,
116, 183-185,
187 |
|
|
|
| FIGURE 7.1 Drug Metabolism
by CYP Enzymes |
 |
| Modified from reference:
282 |
|
Using this information, a classification system has been
developed based on the degree of structural similarities between
different enzymes (ie, amino acid sequence homology).187
The rationale for such a classification system is that the
more similar the structure, the closer the enzymes are both
phylogenetically and functionally (ie, function follows structure).
The CYP enzymes are grouped into families designated by the
first number. All enzymes in a family have at least 40% amino
acid sequence homology. They are further grouped into subfamilies
designated by an alphabet letter. All enzymes in the same
subfamily have at least 55% amino acid sequence homology.
The last number designates the gene that codes for a specific
enzyme. Table 7.3 shows all of the human
CYP enzymes grouped into families and subfamilies.
These enzymes are divided into two major groups (Table
7.4):
- "Steroidogenic" CYP enzymes
- "Xenobiotic" CYP enzymes
The former are phylogenetically older, occurring in
even single cell organisms. They are located in the mitochondria
of cells and are responsible for the synthesis
of steroids and other substances necessary for the maintenance
of cell wall integrity (Table 7.5).
Substantial impairment in the functional integrity of the
steroidogenic enzymes (eg,
deficiency due to genetic mutation) is incompatible with life.
The xenobiotic CYP enzymes evolved from the steroidogenic
CYP enzymes over 1 billion years ago (Table
7.4). These enzymes are located in the smooth endoplasmic
reticulum of cells (Figure 7.1) and
appear to have evolved during the era of plant-animal differentiation.107,186
The term "xenobiotic" refers to the ability of these
enzymes to metabolize foreign (ie, xeno) biological substances.
These enzymes allowed animals who possessed them to metabolize
plant toxins before they could enter the animal's systemic
circulation and cause damage. Thus, these enzymes conveyed
survival advantage to the animals that possessed them.
Since drugs originally came from plants and resemble plant
toxins, they can be metabolized by the same enzymes. In fact,
these enzymes determine in part what substances can become
drugs. If a mechanism did not exist to eliminate a substance,
then that substance could not be used
as a drug.
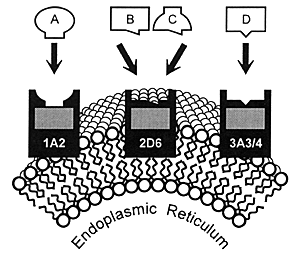
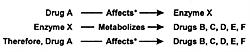
As illustrated in Figure 7.1, many
drugs have a principal CYP enzyme that is responsible for
the bulk, if not all, of its metabolism. In this illustration,
Drug A is principally metabolized by CYP 1A2, Drug B by CYP
2D6, and Drug D by CYP 3A3/4. Drug C is an example of a drug
that can be metabolized by either CYP 2D6 or 1A2.
This latter situation has caused some confusion in that
it has been erroneously stated that "another enzyme could
take over for an inhibited enzyme." However, these enzymes
do not change their enzymatic activity or affinity for a substrate
based on whether the principal enzyme is inhibited. Instead,
a second enzyme comes into play when the concentration of
a substrate (eg, drug) such as Drug C has
reached a sufficient level in the body that this pathway becomes
meaningful for biotransformation and subsequent elimination.
The genetic material for these enzymes is carried in every
cell in the body and expressed in multiple cells in the body.
For example, CYP 2D6 is found in the brain where it is spatially
linked to the dopamine uptake transporter pump.139
However, our knowledge of the precise role of these enzymes
in organs other than the liver and the bowel wall remains
rudimentary.147
In the liver and bowel wall, these enzymes are responsible
for the bulk of phase I or oxidative metabolism of xenobiotics
including dietary toxins, carcinogens, mutagens, and more
recently, drugs. As mentioned above, their existence has permitted
the development and use of medications in the treatment of
patients (Table 7.5).
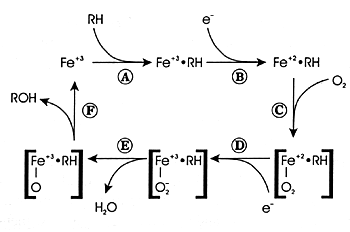
What Is Oxidative Metabolism?
Oxidative metabolism involves the conversion of a substance
into a more polar
species (ie, a metabolite) by the insertion or incorporation
of atmospheric oxygen into the molecule (Figure
7.2). In some instances, the oxidized product is the final
metabolite. In other instances, it is an intermediate metabolite
and undergoes further biotransformation. CYP enzymes mediate
a number of different biochemical reactions (Figure
7.3). The final result of each of these biotransformations
is to produce a polar metabolite that can be eliminated in
urine or feces. Often the final step is conjugation of the
metabolite at a polar site with a moiety such as glucuronic
acid (ie, phase II metabolism). The resulting conjugated product
is water soluble and can be eliminated in the urine. This
sequence from phase I to phase II metabolism and eventual
elimination is illustrated in Figure 7.4.
Why Do We Have Such Enzymes?
| FIGURE 7.2 CYP Enzyme
Reaction Cycle |
 |
| Reference: 180 |
|
| |
|
|
| FIGURE 7.3 Diverse Monooxygenase
Activities of CYP Enzymes |
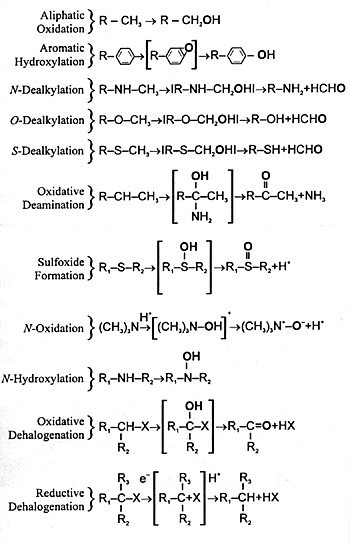 |
| The oxygen derived from
atmospheric oxygen is denoted by boldface. |
|
As mentioned above, the development
of specific CYP enzymes had, at one time, survival value for
animals by metabolizing dietary toxins before they could enter
the systemic circulation. Whether these enzymes still serve
such a role for man is unclear. By facilitating the biotransformation
of dietary toxins, carcinogens and mutagens, these enzymes
determine the total cumulative exposure to such agents in
terms of both absolute concentration achieved and duration
of exposure once such chemical entities are ingested.114,117
This fact has raised the possibility that deficiency in these
enzymes might be a risk factor in the development of specific
diseases caused by environmental exposure to such chemicals.53-55
For example, an active area of research is whether cigarette-induced
increase in the functional activity of CYP 1A2 may explain
the increased risk of some cancers in cigarette smokers;163,181
CYP 1A1/2 transforms some procarcinogens into carcinogens.50,121,193
Another example is the theory that individuals genetically
deficient in CYP 2D6 may have an increased
risk of early-onset Parkinson's disease because they lacked
this enzymatic barrier in the liver to the entry of an environmental
dopamine neurotoxin such as MPTP.17,91
The primary approach in this area of research has been to
study the relative risk of different illnesses as a function
of genetically determined deficiency in specific enzymes.54
What Is the Role of These Enzymes
in Determining Response to Drug Therapy?
The functional state of these enzymes is the major determinant
of the rate of biotransformation of the drug into metabolites
and, hence, a major determinant of the elimination rate (ie,
clearance) of specific drugs. As such, their activity determines
the concentration of the drug and/or its metabolites that
will be achieved in the body as a function of the dose administered
(Figure 7.5). Their activity also determines
the relative ratio of the administered drug to its metabolites.
This ratio may also be important because
the metabolite(s) may be biologically active and that activity
may differ in clinically important ways from that of the parent
drug (eg, clomipramine and desmethylclomipramine)
(see Section 3).
| FIGURE 7.4
Phases of Xenobiotic Metabolism |
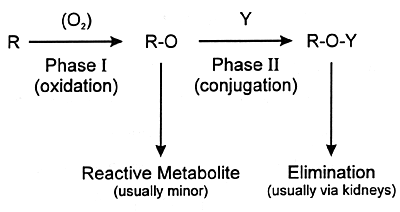 |
| Y = moiety such as glucuronic
acid |
|
| |
|
|
| FIGURE 7.5
Relationship Between Dosing Rate, Clearance, Steady-state
Drug Concentration and Clinical Response |
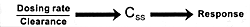 |
| CSS = Steady-state
drug concentration |
|
To put this matter in perspective, recall that the concentration(s)
of the parent drug and/or its metabolites is one of the 3
variables that determines the magnitude and/or the nature
of the patient's response to the drug treatment (Figure
7.6). Obviously, the drug must affect a site of action
(SOA) in a specific way (ie, act as an agonist or antagonist
at the SOA) to produce the desired effect. However, the magnitude
of the effect is dependent on the concentration of the drug
achieved at that SOA. In addition to these 2 variables, biological
variance among different patients can shift the dose-response
(ie, concentration-response) curve. For example, a drug such
as propanolol slows heart rate by its negative chronotropic
effect mediated by its mechanism of action (MOA) (ie, the
blockade of B-adrenergic receptors). The higher the
concentration of propanolol, the greater the degree of B-adrenergic
receptor blockade, and the slower the heart rate. A patient
with an underlying cardiac disease may experience a substantially
greater effect at the same concentration (ie, their dose-
or concentration-response curve has been shifted to the left
due to their underlying cardiac condition).
| FIGURE 7.6 Relationship
of Pharmacodynamics, Pharmacokinetics and Biological Variance
in Determining Overall Result of Drug Treatment |
 |
Since the magnitude and nature of the patient's response
is concentration-dependent, the functional activity of these
enzymes can make the difference between therapeutic success
and failure. Such therapeutic failure may take the form of
either suboptimal response or toxicity. A suboptimal response
can result from the development of an inadequate concentration
at the SOA due to unusually rapid clearance. Toxicity can
result from the development of excessively high concentrations
due to unusually slow clearance. In essence, a change in the
elimination rate (ie, clearance) of a drug is the mirror image
of a change in its dosing rate (Figure 7.5).
Parenthetically, clearance is the amount of drug cleared per
unit of time. The reason is the concentration of drug achieved
in the body is directly proportional to the dosing rate and
is inversely proportional to its elimination rate.
This inverse relationship is illustrated in Figure
7.5. Due to this relationship, a change in the functional
activity of the enzymes mediating the biotransformation of
a drug necessary for its eventual elimination typically produces
an effect the opposite of what would occur with a change in
the dosing rate. For example, a decrease in functional activity
in the CYP enzymes will lead to a decrease in drug clearance
and an increase in drug accumulation (ie, the same net effect
as will occur with a dose increase). Conversely, an increase
in functional activity of the enzyme would typically result
in a decrease in drug accumulation (ie, a net effect similar
to a dose decrease).
How Do These Enzymes Relate to
Drug-Drug Interactions?
Drugs can increase or decrease the functional activity of
CYP enzymes. Induction of the gene responsible for
the production of the enzyme increases its rate of synthesis,
thus increasing the cellular content and activity of the induced
CYP enzyme.145,206
Since induction involves protein synthesis, there is a delay
in both its onset and its offset relative to starting and
stopping the inducer. Therefore, the full effect may not be
apparent for several weeks after the inducer has been started
and the effect will take a similar period of time to fully
dissipate after the inducer has been stopped and the rate
of enzyme production has returned to baseline.
Drug-induced inhibition is usually competitive and occurs
immediately.278
However, the magnitude of the inhibition is a function of
the concentration of the inhibitor (see Section
8). Thus, the half-life of the inhibitor will determine
how long it must be administered before the full effect is
achieved and, conversely, how long after its discontinuation
the inhibition will persist. For this reason the full effect
of fluoxetine, and particularly its long-lived active metabolite
norfluoxetine, on several CYP enzymes can take several weeks
to be achieved and can persist for several weeks after its
discontinuation.112,219
How Do Such Drug-Drug Interactions
Present Clinically?
| TABLE
7.6 Types of Drug Interactions |
| Pharmacodynamic |
Mechanisms of action of one drug
amplifies or diminishes the effect produced by mechanisms
of action of another drug. |
| Pharmacokinetic |
Effect of one drug alters the
pharmacokinetics of another, leading to a change in its
effective concentration at its site(s) of action. |
| Data
from reference: 213 |
There are two types of drug-drug interactions: pharmacodynamic
and pharmacokinetic (Table 7.6).224
In the former, both drugs are acting via their MOA to produce
a physiological response and where the effect of one drug
on its SOA amplifies or diminishes the response produced by
the effect of the other drug on its SOA. For this reason,
a pharmacodynamic drug interaction is likely to produce a
qualitative change in the patient's response (ie, a change
in the nature of the response). Examples of a qualitative
change include the central serotonin syndrome that can occur
when an SSRI and a monoamine oxidase inhibitor (MAOI) are
taken together. Due to the frequently dramatic presentations
of pharmacodynamic drug interactions, they are often readily
identified and, hence, have received considerable attention
in the medical literature.
In contrast, pharmacokinetic interactions are more likely
to present as a quantitative (ie, more or less) rather than
a qualitative change in the patient's response (Table
7.7). These types of interactions occur when the second
drug affects the pharmacokinetics of the first drug and alters
the concentration of the first drug at its SOA. In contrast
to pharmacodynamic drug interactions, these interactions do
not involve the addition of another effect on another SOA
to the treatment equation. For this reason, a pharmacokinetic
drug interaction often produces the same result as would occur
with a dose change and, thus, is something that can be expected
when only using the affected drug. In other words, the result
is a known effect of the drug whose pharmacokinetics were
altered by the addition
of a second drug.
| TABLE 7.7 Pharmacokinetic
Interactions: How Do They Present Clinically? |
- Generally quantitatively (ie, magnitude of
the response) rather than qualitatively (ie,
nature of the response)
- Rather than an unexpected response, such
an interaction typically produces an expected
response but at an unexpected dose (eg, failure
to respond, adverse effect)
- This outcome is easily attributed to the
patient being either sensitive or resistant
to the affected drug
- May also present as an erratic response which
can be dismissed as being due to noncompliance
|
|
|
| FIGURE 7.7 Dose-Response
Curves with Risperidone |
 |
| Extrapyramidal Adverse
Effects |
 |
| Data from reference: 169 |
|
| |
| FIGURE 7.8 Dose-response
Curves for Seizure Risk |
| Clozapine1 |
 |
| Bupropion2 |
 |
| Data from references: 160,
272 |
|
However, the effect is occurring at an unexpected dose due
to the change in the drug's clearance. What the physician
may not realize is that adding the second drug has, in essence,
changed the effective dose of the first drug by altering its
clearance (Figure 7.5). Thus, the patient
may appear unusually sensitive to the first drug (ie, develop
a known dose-dependent adverse effect of the first drug on
a dose that is usually well tolerated) or nonresponsive to
the first drug (ie, failure to optimally respond to what is
usually an effective dose). Thus, pharmacokinetic drug interactions
may be dismissed as being due to idiosyncrasies on the part
of the patient rather than being recognized for what they
are.
Some examples may be useful to illustrate this concept.
Risperidone is a recently marketed antipsychotic with some
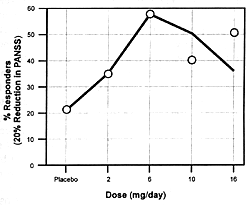
unique features. Figure 7.7 shows the
dose-dependent nature of its antipsychotic efficacy and its
risk of causing extrapyramidal adverse effects. As the dose
of risperidone goes up, there
is initially an increase in its antipsychotic efficacy; but
somewhere between 8 to 10 mg/day, its antipsychotic efficacy
either plateaus or actually begins to decrease.169
However, the incidence and severity of extrapyramidal adverse
effects continue to go up with dose increases. For these reasons,
6 mg/day is the recommended optimal dose. If a concomitantly
administered drug decreases the clearance
of risperidone, the consequence can be the same as increasing
its dose. The result will be a reduction in its efficacy and
an increase in the incidence of extrapyramidal adverse effects.
If the clinician does not know the dose had been effectively
changed by adding the inhibitor, s/he may simply conclude
that the patient is resistant to the beneficial effects and
sensitive to the adverse effects of risperidone. This drug
is metabolized by the CYP enzyme 2D6.
The consequences can also be more serious than loss of efficacy
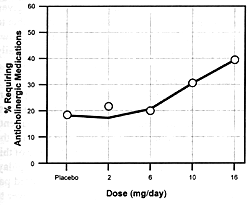
and increased extrapyramidal side effects. Figure
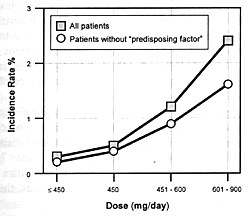
7.8 shows the seizure risk due to clozapine as a function
of its daily dose. If a drug inhibits the clearance of clozapine,
the consequences will be similar to increasing its dose. If
the patient is on 300 mg/day and the clearance rate is reduced
by 50%, this will be comparable to doubling the dose. With
such a functional dose increase, the patient's risk of experiencing
a seizure will go from 1.5% to 5.0% (ie, over a threefold
increase in risk).
Even though such an increase in the relative risk of this
potentially serious adverse effect for a given patient is
substantial, it will be virtually impossible to detect in
clinical practice. Since the clinician knows that some patients
will have seizures even on daily doses of 300 mg/day, s/he
may simply conclude that the patient is "sensitive"
to this adverse effect.
To prove that such a pharmacokinetic drug interaction causes
the increased risk by functionally increasing the dose would
require a formal study of patients randomly assigned to clozapine
with and without the inhibitor. Given the relatively infrequent
nature of this adverse effect, even at doses of 600 to 900
mg/day, the study would have to involve several hundred patients
to be assured of having the statistical power to detect the
difference in the predicted seizure risk for the two conditions
(ie, 1.5% risk versus a 5% risk). Such a study will likely
never be done for ethical and economic
reasons. Hence, physicians will need to make decisions based
on an understanding of these principles. Of note, several
CYP enzymes have been implicated in the metabolism of clozapine,
including CYP 1A2, 3A3/4208and
possibly 2D6,255
although there is conflicting data for this CYP enzyme. Also,
fluvoxamine and fluoxetine have been reported to cause moderate
increases in clozapine levels,137,300
but it is not known which enzymes they are affecting
to produce this increase.
Bupropion, which is an effective antidepressant with several
advantageous features,214,220
has a dose-dependent risk of seizures (Figure
7.8). For that reason, this drug has only been marketed
in the U.S.; nonetheless, it provides another good illustration
of this point. Bupropion undergoes extensive biotransformation
to active metabolites, but the enzymes mediating those transformations
have not been established.214
Inhibition of bupropion's clearance or the clearance of its
active metabolites can increase the seizure risk just as occurs
with a dose increase. Limited case data indicate that fluoxetine
can produce substantial elevation of two metabolites of bupropion,
although the precise mechanism for this effect has not been
established.214
Again, a large scale study of hundreds of patients would be
needed to statistically test the hypothesized increased seizure
risk with such a combination.
Despite the general rule being quantitative changes in response,
pharmacokinetic drug interactions can sometimes produce a
qualitative change in the patient's response. Two examples
are provided in Table 7.8. Since qualitative
changes are easier to recognize in clinical practice than
are quantitative changes, these interactions are better known.
In the first type, the drug whose clearance is changed has
effects on multiple SOAs in the body in a concentration-dependent
manner.221,225
Tricyclic antidepressants (TCAs) are excellent
examples (Figure 7.9). At low concentrations,
they block histamine and acetylcholine receptors, producing
effects mediated by those actions. At therapeutic concentrations,
TCAs inhibit the norepinephrine and serotonin uptake pumps
believed to mediate their antidepressant efficacy. At toxic
concentrations, TCAs inhibit fast sodium channels; hence,
they can slow intracardiac conduction leading to serious,
and even fatal, conduction disturbances. This same MOA is
probably responsible for their central nervous system (CNS)
toxicity (ie, delirium, seizures and coma).
If a drug inhibits the clearance of TCAs, it will have the
same effect as increasing the dose of the TCA. The response
to such a pharmacokinetic drug interaction may be a fatality
rather than an antidepressant response.216
That is what is meant by a change in the quality (ie, nature)
of the response (eg, seizure or arrhythmia, as opposed to
antidepressant efficacy) as opposed to the quantity of the
response (eg, severity of dry mouth). The principal enzyme
responsible for the clearance of TCAs particularly secondary
amine TCAs (eg, desipramine, nortriptyline) is the CYP enzyme
2D6 which mediates the ring hydroxylation of these drugs.
This conclusion is based on extensive and varied types of
pharmacokinetic data.25,40,42,45,46,48,147,195,223,262
The N-demethylation pathway, which is more prominent
for tertiary amine TCAs (eg, amitriptyline, imipramine) than
for secondary amine TCAs, although still not the principal
route of elimination, is mediated by several CYP enzymes including
CYP 1A2, 2C19 and 3A3/4.42,46,58,161,168,192,202,256,257
Pharmacokinetic drug interactions can also present qualitatively
when the inhibition of the enzyme significantly changes the
ratio of the parent drug to a metabolite which is pharmacologically
active in a substantially different way than the parent drug.
An example is the inhibition of the conversion of terfenadine
to its acid metabolite. That conversion
is mediated by CYP 3A3/4 and occurs in the bowel wall and
liver during the absorption of terfenadine.295
This conversion is typically so efficient and complete that
virtually all of the terfenadine is converted to its acid
metabolite before entry into the systemic circulation. This
metabolite is an active antihistaminic agent and is relatively
devoid of effects on intracardiac conduction. Therefore, the
use of terfenadine generally does not carry a risk of adverse
effects on intracardiac conduction. However, terfenadine itself
does inhibit intracardiac conduction and can cause arrhythmias.258
If the conversion of terfenadine to its acid metabolite is
substantially blocked during absorption, then terfenadine
will enter the systemic circulation. If its concentrations
are sufficiently high, cardiac arrhythmias can result. Thus,
coadministration of terfenadine with a potent inhibitor of
CYP 3A3/4 can have serious, and even life-threatening, adverse
consequences.297
Ketoconazole related antifungal agents and several macrolide
antibiotics (eg, erythromycin) can substantially inhibit this
enzyme.29,126,127,279
Fluvoxamine, norfluoxetine and the non-SSRI antidepressant,
nefazodone, also inhibit this enzyme, but are substantially
less potent than ketoconazole
in this regard.144,276
| TABLE 7.8
Examples of Metabolically Mediated Pharmacokinetic
Drug-drug Interactions and Their Typical Clinical
Presentations* |
| Type |
Presentation |
Affected
Drug |
Causative
Drug |
CYP Enzyme |
| Buildup of drug levels
due to inhibition of clearance |
Increase in incidence and/or
severity of expected dose-dependent adverse effects |
Carbamazepine
Dextromethorphan
Phenytoin
Propanolol
Theophylline
Tricyclic antidepressants
Tricyclic antidepressants
Warfarin
|
Erythromycin
Paroxetine
Fluoxetine
Cimetidine
Fluvoxamine
Fluoxetine
Fluvoxamine
Fluvoxamine
|
3A3/4
2D6
2C9/10
1A2, 3A3/4
1A2
2D6
1A2, 3A3/4
1A2 |
| Reduction of drug levels
due to induction of clearance |
Loss of efficacy |
Disopyramide
Oral contraceptives
Theophylline
Propanolol
Quinidine
Theophylline
Warfarin
|
Phenytoin
Carbamazepine
Phenytoin
Rifampin
Phenobarbital
Phenytoin
Secobarbital |
3A3/4
3A3/4
1A2
3A3/4
3A3/4
1A2
1A2 |
| Blockade of the production
of an active metabolite |
Loss of efficacy |
Codeine |
Paroxetine |
2D6 |
| Increased accumulation
of an unusual toxic parent drug or metabolite |
Unexpected toxicity
based on the usual
pharmacology of the drug |
Astemizole
Terfenadine |
Ketoconazole |
3A3/4 |
| * The above
listing is in no way an exhaustive list of causative
or affected drugs for either a type of interaction
or for a specific CYP enzyme, but simply representative
examples. |
| Reference:
224 |
|
|
|
| FIGURE 7.9 Multiple Concentration:
Response Curves of Tertiary TCAs |

|
| Reference:
221 |
|
Where Are We Going With This Knowledge?
| FIGURE 7.10
How Knowledge of Drug-metabolizing Enzymes Will Simplify
Understanding of Pharmacokinetic Interactions |
 |
| * Could be inhibition or induction. |
Two sets of knowledge are now being developed as illustrated
in Figure 7.10. The first is
which drugs affect which CYP enzymes, either by induction
or inhibition. The second is which drugs are metabolized by
a specific CYP enzyme (Table 7.9). With
these two sets of knowledge, we can determine whether the
coadministration of one drug is likely to affect the biotransformation
of another drug. We can also determine whether the second
drug is likely to alter the overall clearance (ie, elimination
rate) of the first drug and hence its accumulation on a given
dose and whether the second drug is likely to affect the relative
ratio of specific metabolites to the parent drug by inducing
or inhibiting a specific biotransformation pathway. In addition
to the examples already given, the coadministration of an
inhibitor or an inducer might increase the production of what
is normally a minor metabolite to a substantial extent. This
metabolite may mediate clinically important effects. For example,
the increased risk of hepatoxicity when valproate is coadministered
with another anticonvulsant (ie, carbamazepine or phenytoin)
may have been due to the increased production of the 4-ene
metabolite of valproate via the induction of the CYP enzyme
that mediates the conversion.169
| TABLE 7.9Drugs Metabolized
by CYP Enzymes* |
| CYP 1A2 |
Antidepressantsamitriptyline,
clomipramine, imipramine Antipsychoticsclozapine
B-Blockerspropanolol Miscellaneouscaffeine,
paracetamol, theophylline, R-warfarin |
| CYP 2C9/10 |
| phenytoin, S-warfarin,
tolbutamide |
| CYP 2C19 |
Antidepressantscitalopram,
clomipramine, imipramine
Barbiturateshexobarbital, mephobarbital,
S-mephenytoin
Benzodiazepinesdiazepam
B-Blockerspropranolol |
| CYP 2D6 |
Antiarrhythmicsencainide,
flecainide, mexiletine, propafenone Antipsychoticshaloperidol,
perphenazine, risperidone, thioridazine
B-Blockersalprenolol, bufarolol, metoprolol,
propranolol, timolol Miscellaneousdebrisoquin,
4-hydroamphetamine, perhexiline, phenformin, sparteine
Opiatescodeine, dextromethorphan, ethylmorphine
SSRIsfluoxetine, N-desmethylcitalopram,
paroxetine
TCAsamitriptyline, clomipramine, desipramine,
imipramine, N-desmethylclomipramine, clomipramine,
nortriptyline, trimipramine
Other Antidepressantsvenlafaxine, the mCPP
metabolite of nefazodone and trazodone |
| CYP 3A3/4 |
Analgesicsacetaminophen,
alfentanil, codeine, dextromethorphan Antiarrhythmicsamiodarone,
disopyramide, lidocaine, propafenone, quinidine
Anticonvulsantscarbamazepine, ethosuximde
Antidepressantsamitriptyline, clomipramine,
imipramine, nefazodone, sertraline, O-desmethylvenlafaxine
Antiestrogensdocetaxel, paclitaxel, tamoxifen
Antihistaminesastemizole, loratadine, terfenadine
Antipsychoticsclozapine Benzodiazepinesalprazolam,
clonazepam, diazepam, midazolam, triazolam
Calcium Channel Blockersdiltiazem, felodipine,
nicardipine, nifedipine, niludipine, minodipine,
nisoldipine, nitrendipine, verapamil Immunosuppressantscyclosporine,
tacrolimus (FK506macrolide)
Local Anestheticscocaine, lidocaine Macrolide
Antibioticsclarithromycin, erythromycin, triacetyloleandomycin
Steroidsandrostendione, cortisol, dihydroepiandrosterone
3-sulfate, dexamethasone, estradiol, ethinylestradiol,
progesterone, testosterone Miscellaneousbenzphetamine,
cisapride, dapsone, lovastatin, omeprazole (sulfonation) |
| * Major pathway for elimination
of tricylcic antidepressants is ring hydroxylation;
N-desmethylation, a minor pathway, is mediated
by several CYP enzymes. |
| Adapted from references:
39, 119,
144 |
|
|
|
| TABLE
7.10 Genetically Determined CYP Enzyme Deficiency |
| CYP |
Orientals |
Blacks |
Whites |
| 2C19 |
20% |
Not
known |
3% |
| 2D6 |
1% |
4%
|
8% |
| Data from references:
104, 152 |
|
In the past, this type of pharmacokinetic drug interaction
could
not be anticipated, but had to occur and then be identified
clinically after the fact. Early pharmacokinetic drug interaction
studies were done mainly on the basis of what drugs were likely
to be used together rather than because there was a substantial
reason to suspect a clinically meaningful drug-drug interaction.
Physicians had to remember these types of interactions without
having any organizing principles to aid them.
Due to improved knowledge of CYP enzymes, this situation
has substantially changed. Now studies can be done by deductive
reasoning using the 3 sets of information illustrated in Figure
7.10. From a research standpoint, this knowledge will
permit the anticipation of clinically meaningful interactions
which can then be confirmed by focused formal pharmacokinetic
drug interactions studies done under clinically relevant conditions.
From a clinical standpoint, this knowledge will allow physicians
to anticipate such an interaction and make appropriate drug
and dose selections to avoid undesired consequences.
This knowledge is also being used to determine what drugs
to develop. New candidate drugs are now tested for how they
are metabolized and whether they affect specific CYP enzymes.
A new candidate drug may not be developed based on the results
of such tests if it is principally metabolized by a CYP enzyme
that is inducible or inhibited by many commonly used drugs
or is genetically deficient in a sizable percentage of the
population (Table 7.10). Also, a new
drug may not be developed if it induces or inhibits CYP enzymes
responsible for the metabolism of many other drugs.
If the unique efficacy of the new drug outweighs these considerations,
then the information about how it is metabolized and its affects
on the CYP enzymes will be used to help guide its safe and
effective use in clinical practice. For example, this information
can be entered into a readily accessible computer database;
the physician can enter the patient's current treatment into
a computer and the new drug which is going to be added to
the regimen. Using such an information system, the physician
can quickly determine:
- Whether an interaction is likely
- The nature of the interaction, including its potential
seriousness
- What adjustment should be made in terms of dosage
- What drug can be substituted for the proposed new drug
to avoid the interaction
Patients can also be genotyped and phenotyped for specific
CYP enzyme activity, and that information can also be entered
into the computer database along with their current treatment
regimen and any proposed changes. This additional information
will further increase the accuracy of the predictions and
recommendations. Obviously, the development of this knowledge
has substantial implications for improving patient care in
terms of safety, tolerability and efficacy. Those implications
extend well beyond any single class of medications, but nonetheless,
have specific implications now for the SSRI class of antidepressants
as discussed in Section 8.
|



