|
|
| De-Spinning In Vitro Data |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, September 1999, 283-287
|
"Spin" refers to framing events in a way that is favorable to the person doing the "spinning." It is a term that becomes commonplace during political campaigns. Candidates running for major office have one or more "spin doctors" who specialize in putting a "positive spin" on events affecting their candidate and a "negative spin" on events affecting their opponents. While generally not a lie, "spin" may nevertheless mislead the listener.
For example, during the height of the Cold War, a Soviet news anchorman reported the results of an important qualifying race as follows: The Soviet runner came in second and the American runner came in next to last. What was not mentioned in the report was that it was a two-man race! This report, although factually accurate, was nevertheless misleading. That is spin. In the same way, an attorney might appeal to the jury for leniency since his clients are orphans after they murdered their parents.
So what does "spin" have to do with in vitro psychopharmacology?
Companies naturally endeavor to portray their products in the most favorable light possible. Sometimes, they can do this using compelling, straightforward data. At other times, however, they have to rely on "spin." For that reason, clinicians need to be able to critically examine the basis for promotional claims. The goal of this column and the others in this series is to present a way of critically thinking about the potential relevance of in vitro data to actual clinical use of a drug.
The meaning of in vitro pharmacology is easily "spun" for several reasons. First, head-to-head in vitro data for different drugs can be generated much faster and at much less cost than head-to-head clinical data. You can find tables comparing the in vitro binding affinity of a host of drugs for a variety of different receptors. Second, clinicians often assume that a difference between two drugs in binding affinity for a receptor must be clinically relevant. One can thus imply a clinically significant difference to the unsuspecting based solely on the fact that a drug has a higher or lower affinity for a given target than the competitor's product.
While differences in binding affinity can be quite important clinically, this is not always the case. Binding affinity is only one factor in the equation:
(Equation 1)
Effect = potency at x drug x biological
site of action level variance
Binding affinity has two major implications for the clinical use of a drug. First, it tells you which receptor a drug will affect first (i.e., at its lowest concentration). Second, it tells you what concentration of the drug is needed to affect a specific target. Drugs A and B may affect the same target equally at their respective usual therapeutic doses even though drug A has a 10-fold higher binding affinity for the target than drug B. The reason is that the concentration of drug B at its therapeutic dose is 10 times higher than drug A at its therapeutic dose. In fact, the binding affinity of the drug for the target mediating a specific desired effect determines the concentration (and hence the dose) of the drug needed to produce that effect. Recall the second equation:
(Equation 2)
Concentration = dosing rate / clearance
In my last column, I discussed the issue of selectivity as it relates to antidepressants. In this column, I continue that discussion and present the binding profile of all the members of the SSRI class of antidepressants. I will also address some of the specific questions posed by colleagues around the country that inspired this series of columns. These questions all revolve around the issue of whether differences in the in vitro binding affinity of different antidepressants have any physiological/clinical implications. For example, colleagues have asked:
- Is citalopram the most "selective" SSRI?
- Is sertraline an inhibitor of dopamine uptake?
- Is paroxetine a blocker of muscarinic acetylcholinereceptors?
As I mentioned in the last column, the answer to these questions depends on what you mean by "is." In the first question, it depends on how you define the gap (i.e., what is your starting point and your ending point?). In the other two questions, it depends on whether you mean what is theoretically possible, given the in vitro affinities, or what is most likely to actually occur in the clinically relevant dosing range.
Potency is a relative concept that involves comparing the binding affinities of different drugs for the same target (e.g., which one is the most potent serotonin uptake inhibitor). Alternatively, one can compare a single drug's affinity for different targets (e.g., whether Drug A is more potent as a serotonin than as a norepinephrine uptake inhibitor). Either way, the binding affinity tells you how much drug is needed to affect a target to a physiologically meaningful degree. In other words, knowing the potency of the drug for a target in an individual and knowing its concentration in that individual allows you to calculate the expected occupancy of that target by that drug in that individual. This is the basic rationale for therapeutic drug monitoring (i.e., to estimate which targets a drug occupies and to what extent in a given patient).
Why are SSRIs so alike?
Except for clinically relevant differences in their pharmacokinetics and their effects on oxidative drug metabolism, all of the SSRIs are quite similar to each other (Table 1). That is why the differences in their pharmacokinetics and their effects on oxidative drug metabolism take on such importance when evaluating the relative merits of these drugs.1,2 If there were clinically meaningful differences in the neuropsychopharmacology of the various SSRIs, then the differences in their pharmacokinetics and effects on oxidative drug metabolism would be less pivotal as the defining difference among the members of this class.
SSRIs have so much in common because each was developed with the intent of producing a molecule capable of selectively binding to and blocking the serotonin uptake pump. That goal was achieved, as can readily be seen in Figure 1, which shows the binding affinities of the five currently marketed SSRIs for a number of clinically important neural mechanisms of action. The IC50 for binding to the serotonin uptake pump for each of these drugs is at least 10-fold greater than for binding to any of the other neural mechanisms shown in Figure 1.
| Table 1 - The pharmacologic properties shared by the SSRIs1,2 | - Equivalent acute and maintenance antidepressant efficacy
- Flat dose-response curve in terms of antidepressant efficacy
- Ascending dose-response curve in terms of adverse affects
- Similar adverse effects profile consistent with excessive serotonin agonism
- All produce 60% - 80% inhibition of serotonin uptake inhibition at their loweest, usually effective antidepressant dose.
- Efficacy in several anxiety syndromes as well as major depression
|
As I discussed in the last column, a 10-fold difference in a drug's binding affinity for two targets means that a concentration of the drug can be achieved that produces physiologically meaningful occupancy (and hence effect) of the high affinity target but virtually no occupancy (and hence no effect) of the second, lower affinity target.
Figure 1 shows you why the SSRIs are called "serotonin selective" and why their neuropsychopharmacology is so similar in terms of efficacy and adverse effects. They were, after all, built that way. This is why one would predict that switching among the SSRIs in the case of nonresponse would be of value only if the patient did not achieve a therapeutic concentration of the first SSRI tried. The SSRIs do differ in the cytochrome P450 (CYP) enzymes that metabolize them. If a rigorously designed study demonstrated that SSRI 2 worked when SSRI 1 did not, despite achieving usual therapeutic concentrations of both, then that would indicate that there are differences in mechanism of action (e.g., receptor or enzyme) between the two and that we should search for those differences. However, the studies that have been done to date on switching among the SSRIs are so flawed as to be virtually worthless.4
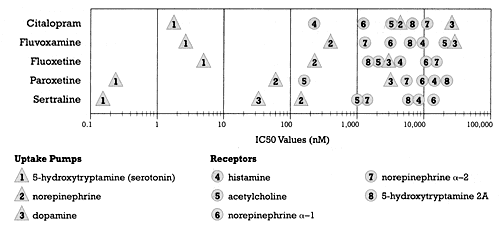 | Figure 1 - Relative Potency for Different Sites of Action for the SSRI Class of Antidepressants
- Based on data from Hyttel 1993 |
Is citalopram the most "selective" SSRI?
This claim has been made on the basis of in vitro data. What is the "spin?" If you define "selective" to mean the difference in the binding affinity of the SSRI for the serotonin versus the norepinephrine uptake pump, then citalopram is the most "selective" as shown in Figure 2.3 Citalopram is 3,400 times (i.e., over three orders of magnitude) more potent at inhibiting the serotonin than the norepinephrine uptake pump. See my last column, "Defining `Is'," to put this difference in perspective in terms of what it means for serotonin versus norepinephrineuptake inhibition. While this difference is factually accurate, the claim that citalopram is the most selective SSRI is nevertheless "spin" for two reasons.
First, this claim ignores the fact that the second target affected by citalopram is not the norepinephrine but rather the histamine receptor (Figure 1). If you define "selective" as the separation between the drug's first and second targets, then both paroxetine and sertraline are more "selective" than citalopram. It is of interest that fluoxetine is the least "selective" of all the SSRIs, with a 10-fold difference in binding affinity between its first and second neural targets (i.e., the serotonin and norepinephrine uptake pumps, respectively).
The second reason rests on the answer to the "So what?" question. If a drug has more than one order of magnitude (i.e., a 10-fold) separation between its effect on its first target and its next target, it can achieve a meaningful effect on the first without affecting the second. Larger gaps in separation may be of interest to the researcher but are of doubtful clinical relevance. Thus, it is unlikely that there will be any clinically meaningful difference in the pharmacology of drugs with two versus three orders of magnitude separation, respectively. But this is not likely to stop someone from touting the greater "selectivity" of his or her drug, a claim that may be factually accurate in terms of in vitro pharmacology but that may have no clinical relevance.
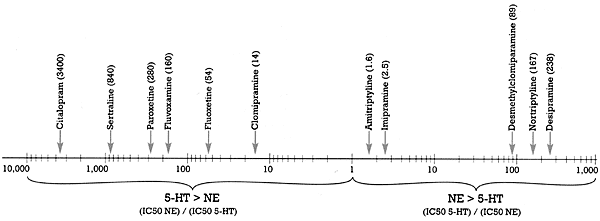 | Drug listing in above diagram (left to right)
Citalopram (3400)
Sertraline (840)
Paroxetine (380)
Fluvoxamine (160)
Fluoxetine (54)
Clomipramine (14)
Amitriptyline (1.6)
Imipramine (2.5)
Desmethylclomiparamine (89)
Nortriptyline (167)
Desipramine (238) | Figure 2 - Selective ratios for a series of Uptake Inhibitors Measured In Vitro
- Based on data from Hyttel 1993 |
Is sertraline an inhibitor of dopamine uptake?
As shown in Figure 1, the second target affected by sertraline is the dopamine uptake pump. In fact, sertraline is considerably more potent at inhibiting this pump than any other SSRI. (In an upcoming column, I will show you that it is also considerably more potent as a dopamine uptake pump inhibitor than is bupropion.)
Given these in vitro data, the pivotal question is "So what?" To answer this, we come back to the magnitude of the separation between sertraline's affinity for its first target, the serotonin uptake pump, and its second target, the dopamine uptake pump. That gap defines the increase in the concentration of sertraline needed to affect the dopamine uptake pump. The affinity of sertraline for the former is 0.19 nM whereas its affinity for the latter is 48 nM.
The importance of in vitro binding is that it tells us what concentration of the drug is needed to achieve meaningful occupancy of a site of action of interest. That in turn is important because the concentration needed determines the dose that is needed to produce the desired effect via the site of action. Thus, the difference in the binding affinity of one drug for two different sites of action tells us what concentration (and hence dose) of the drug is needed to produce meaningful occupancy of those two different sites. To illustrate this point further, let us consider the effect of sertraline on its highest affinity potent neural site of action (i.e., the serotonin uptake pump) versus its second highest affinity neural site of action (i.e., the dopamine uptake pump) (Figure 1).
Of all drugs labeled for use as an antidepressant, sertraline is the most potent in terms of dopamine uptake inhibition. Nevertheless, sertraline is 250 times more potent as an inhibitor of the serotonin uptake pump than it is as an inhibitor of the dopamine uptake pump (Figure 1). As discussed in my last column, that separation in potency means that sertraline can achieve a concentration that produces virtually complete inhibition of the serotonin uptake pump without producing any inhibition of the dopamine uptake pump. That is why sertraline, like the other SSRIs, is called "serotonin selective."
As discussed in an earlier column,6 studies have been done to determine the dose- (and hence concentration-) dependent effect of sertraline on serotonin uptake inhibition using the platelet as a surrogate marker of central nervous system effects. Those studies found that the concentrations achieved on a dose of 50 mg/day produced inhibition near the plateau portion of the curve and that higher doses produced only modestly more inhibition of the pump. Given that finding, it is quite doubtful that sertraline has any meaningful effect on dopamine uptake at concentrations typically achieved at its recommended dosing range, 50-200 mg/day.
This conclusion is further strengthened by the findings in another study7 that examined the concentration-dependent effects of sertraline on platelet serotonin uptake versus the tyramine pressor response, which can be used as an in vivo measure of norepinephrine uptake inhibition. Sertraline produced more than 80% inhibition of serotonin uptake without having any effect on the tyramine pressor response. That is germane to the present discussion because there is less than an order of magnitude separation between sertraline's affinity for the dopamine versus the norepinephrine uptake pump (Figure 1).
While this does not absolutely rule out an effect of sertraline on dopamine uptake at concentrations usually achieved on its recommended dosing range, it places the burden of proof on the proponent of such a theory. Conversely, the in vitro data and these results can be used to estimate the concentration and dose of sertraline that would be needed to affect the dopamine uptake pump (i.e., 250 times the concentration usually achieved on 50 mg/day).
It is of interest that there has been a report of one individual who took 300-400 mg of sertraline several times a day to achieve a psychostimulant effect. That individual may well have been achieving concentrations that produced physiologically meaningful inhibition of dopamine uptake and that is why he experienced a psychostimulant (i.e., amphetamine-like) effect. That individual must also have been quite tolerant of the adverse effects (e.g., nausea, diarrhea) that can occur with supersaturated inhibition of the serotonin uptake pump. That is another reason why selective drugs are selective -- most individuals cannot tolerate a dose that is needed to affect the second target due to adverse effects produced by supersaturating the first site of action.
Is paroxetine a blocker of muscarinic acetylcholine receptors?
Figure 1 shows that paroxetine is unequivocally the most potent blocker of muscarinic acetylcholine receptors of all the SSRIs. Furthermore, its binding affinity for this target is comparable to that of nortriptyline, a tricyclic antidepressant! Again, the question is "So what?"
Most (but not all) in vitro studies have found that paroxetine has the highest affinity for the serotonin uptake pump of all the SSRIs (the Hyttel study is an exception)2. The concentration of paroxetine needed to occupy this target is therefore lower than the concentration needed of any other SSRI. The affinity of paroxetine for the serotonin uptake pump is 0.29 nM, while its affinity for the muscarinic acetylcholine receptor is 210 nM. Just as we discussed with sertraline, you can block muscarinic acetylcholine receptors with paroxetine but you need concentrations (and hence doses) 700 times greater than those needed to block the serotonin uptake pump. The comparison with nortriptyline misses the fact that the separation between nortriptyline's effects on its desired mechanism of action (most likely the norepinephrine uptake pump) and the muscarinic acetylcholine receptor is approximately 20-fold rather than 700-fold. Nevertheless, consistent with its in vitro pharmacology, nortriptyline has a relatively low likelihood of causing anticholinergic adverse effects at doses (and concentrations) that are therapeutic for treating clinical depression compared to tertiary amine tricyclic antidepressants such as amitriptyline.
Conclusion
In vitro data are quite important. They tell us what a drug is capable of doing. They also tell us how much drug is needed to achieve the effect, which in turn tells us what dose is needed. This is the link between pharmacodynamics and pharmacokinetics.
What "take-home" message can the clinician derive from this discussion? First, there is a need to critically examine promotional claims. Second, in vitro data can be used to make informed estimates about what concentration of a drug will be needed to produce a specific effect. Third, the SSRIs, at least relative to the targets shown in Figure 1, are much more alike than they are different. That fact seriously calls into question the practice of switching a patient who has not benefited from one SSRI to another SSRI, unless it is being done for a pharmacokinetic reason.8
In my next column, I will extend the discussion about the clinical relevance of in vitro binding to the dual effect of venlafaxine on serotonin and norepinephrine uptake pumps in terms of both its dose-adverse effects curve and its dose-antidepressant efficacy curve.
References
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Hyttel J, Comparative pharmacology of selective serotonin reuptake inhibitors (SSRIs). Nord J Psychiatry. 1993;47 (suppl 30):5-12
- Preskorn SH, The appearance of knowledge. J Prac Psych Behav Hlth. 1997;3:233-238
- Preskorn SH, Defining "Is." J Pract Psychiatry Behav Health 1999;5:224-8
- Preskorn SH, Finding the signal through the noise: The use of surrogate markers. J Pract Psychiatry Behav Health 1999;5:104-109
- Harvey AT, Preskorn SH, Mechanism of action of venlafaxine in normal male volunteers (Abstract). Clin Pharmacol Ther 1997;61:175
- Preskorn SH, A tale of two patients. J Pract Psychiatry Behav Health 1999;5:160-164
|



