|
|
Clinical Pharmacology of SSRI's
8 - How Does This Knowledge Relate to the Clinical Use of SSRIs? |
|
|
|
|
|
The effect of specific serotonin selective reuptake inhibitors
(SSRIs) on specific cytochrome P450 (CYP) enzymes has been
the major distinguishing factor among these drugs and is clinically
important for three reasons:
- First, these medications are extensively used in the general
population.
- Second, many physicians use these antidepressants, preferentially
in the elderly and medically-ill with major depression due
to their generally benign safety profile (Table
8.1).
- Third, their long-term maintenance use is growing due
to the recognition that major depression is frequently a
recurring illness, and prophylactic treatment with an antidepressant
can decrease the risk of recurrent episodes. During such
prophylactic treatment, the patient may develop an intercurrent
illness (eg, hypertension) and require the addition of another
medication.
For all of these reasons, there is a substantial likelihood
that the patient who is taking an SSRI will be treated with
another medication and hence will be at potential risk for
a drug-drug interaction.
Consistent with these observations, several recent surveys
have shown that many patients being treated with an antidepressant
were also taking other medications and, hence, had the potential
to experience a drug-drug interaction (Table
8.2).
| TABLE 8.1
Prevalence of Major Depression in Specific Medically
Ill Populations |
| Medical Illness |
Prevalence |
| Terminal solid tumors |
25% to 38% |
| Stroke |
27% to 35% |
| Renal disease |
5% to 22% |
| Chronic pain |
35% to > 50% |
| Epilepsy |
20% to 30% |
| Parkinsons disease |
30% to 50% |
| Myocardial infarction |
20% |
| Diabetes mellitus |
10% |
| Data from references:
83, 241,
248 |
|
| TABLE
8.2 Concomitant Use of an Antidepressant With
Other Medications in Different Patient Populations |
| Source |
Population |
Three or
more |
| Holm et al, 19901 |
Outpatients in general
primary care practice |
30% |
| Preskorn et al, 19952 |
Patients in medical school
outpatient psychiatric clinic |
45% |
| Coulehan et al, 19903 |
Outpatients with depressive
symptoms in a medical school primary care clinic |
60% |
| Wolf et al, 19954 |
"Chronic" outpatients
in a Veterans Administration psychiatric clinic
|
60% |
| References:
1125,
2212,
362, 4289
|
|
| TABLE 8.3
Can Inhibition of CYP Enzymes Have Other Consequences? |
- Can be used intentionally
to delay or change the metabolism of another
drug
- Since CYP enzymes such as
CYP 2D6 are found extrahepatically, such inhibition
may have other consequences (either desirable
or undesirable) on organ function other than
the liver
- Are there any long-term consequences
of altering the clearance rate of the natural
substrates for such an enzyme?
|
|
As discussed in Section 3, one
of the goals in the rational development
of the SSRIs was to reduce the likelihood of pharmacodynamic
drug interactions. Originally, the goal was to avoid the multiple
types of pharmacodynamic drug interactions that can occur
when using tricyclic antidepressants (TCAs) in combination
with other drugs due to the multiple mechanisms of action
(MOAs) of TCAs. While SSRIs were developed to avoid this problem,
they were developed before we had the ability to screen for
effects on the CYP enzymes. This fact explains why there are
so many differences among SSRIs with regard to their effects
on these enzymes. In essence, the action of some of the SSRIs
on these enzymes is analogous to the effect of TCAs on fast
sodium channels: these effects are unintended and unnecessary
relative to the desired goal of treating major depression.
Instead of adding to their efficacy, the inhibition of these
enzymes produces the undesired risk of causing pharmacokinetic
drug interactions. This section will focus on the differential
effects of SSRIs on specific CYP enzymes and their potential
for causing such interactions.
A separate issue is whether the inhibition of these enzymes
has any long-term consequences (Table
8.3). This issue, while only theoretical now, is potentially
important since patients may remain on SSRIs for years to
prevent recurrent depressive episodes. For example, prophylactic
treatment with fluoxetine and paroxetine essentially converts
the patient into a phenocopy of genetic deficiency of the
CYP enzyme 2D6 (Table 8.4). Due to the
widespread use of SSRIs for maintenance treatment, a relatively
large segment of the population is being converted into phenocopies
of CYP 2D6 deficiency. Obviously, this enzyme is not essential
to life given the rather widespread (approximately 7%) incidence
of its deficiency in Caucasians. However, it will be important
to know whether functional activity of this enzyme is a risk
factor for the development of chronic illnesses.3,53,55,61,88,138,231
This issue requires specific study as discussed in Section
7.
Myths Concerning SSRIs and CYP
Enzymes
Perhaps as a result of the intense competition among these
drugs, there has been a considerable amount of disinformation
about the issue of CYP enzymes and SSRIs. A number of myths
have arisen that may cause confusion (Table
8.5). Many of these myths involve the effects of these
drugs on the enzyme CYP 2D6, probably because this enzyme
was the first one shown to be substantially inhibited by some
SSRIs. One myth is that the inhibition of these enzymes, particularly
CYP 2D6, is a class issue with SSRIs. A subset of this myth
is that only the in vitro potency of these drugs needs
to be compared to determine their relative clinical effects
without regard to the different concentration of different
SSRIs achieved under clinically relevant conditions. A second
myth is that the inhibition of these enzymes affects only
one or perhaps a couple of drugs. A third myth is that all
the drugs affected by the inhibition of a specific CYP enzyme
are known.
| TABLE 8.4 Some
SSRIs Lack Selectivity With Regard to Effects on Serotonin
Uptake Versus CYP 2D6 |
| SSRI |
Usually Effective, Antidepressant
Dose (mg/day)1 |
Plasma Level (ng/ml)1 |
Serotonin Uptake1 |
CYP 2D62 |
| Citalopram |
40 |
» 85 |
» 60% |
» 15% |
| Fluoxetine |
20 |
» 200 |
» 80% |
» 85% |
| Fluvoxamine |
150 |
» 100 |
»70% |
< 15% |
| Paroxetine |
20 |
» 40 |
» 80% |
» 85% |
| Sertraline |
50 |
»
25 |
»
80% |
»
15% |
1See Table
3.7.
2See Table 8.9. |
In this section, we will address these myths by reviewing
our current knowledge of the effects of the SSRIs on specific
CYP enzymes. We will first review how this information has
been developed so that the reader can understand, and even
anticipate, future developments in our knowledge about the
relative effects of drugs, SSRIs and others, on these enzymes.
While this book is on SSRIs, the issue of drugs affecting
CYP enzymes is not restricted to this class. They are merely
examples of this phenomenon and thus serve to
illustrate a larger point.
| TABLE 8.5 Common Myths
About CYP Enzymes and SSRIs |
- The effect is a class issue
in which all drugs affect the enzyme to the
same extent.
- Effects on CYP 2D6 are responsible
for all the effects of all the SSRIs.
- Other CYP enzymes will take
over for the inhibited enzyme.
- The clinical significance
is limited to a few drugs.
|
|
| TABLE 8.6 Drug-induced
Inhibition of CYP Enzymes |
- Typically competitive inhibition
- Delays clearance of other
substances causing increased and prolonged accumulation
(eg, pharmacokinetic interactions)
- Can be studied both in
vitro and in vivo Questions that
can be answered by these two types of studies:
- In vitro:
- Does a drug have
the potential to inhibit a specific
CYP enzyme?
- If a pharmacokinetic
interaction has been observed, an effect
on what CYP enzyme mediates the interaction?
- In vivo:
- Does an interaction
occur under clinically relevant dosing
conditions?
|
|
How Do We Determine the Effects
of Specific Drugs on Specific CYP Enzymes?
Our knowledge in this area comes from both in vitro
and in vivo studies (Table 8.6).
In vitro studies can be used to determine how potent
a specific drug is as an inhibitor of a specific CYP enzyme.
In vivo studies are done to determine whether the interaction
occurs and to what extent under clinically relevant conditions.
A brief description of the two different approaches follows
as a frame of reference for the remainder
of this section.
In Vitro Studies
Several different techniques can be used in these studies,
including the use of:
- Purified enzyme
- Liver slices
- Human hepatic microsome preparations
- Cells transvected with a specific human CYP gene
These techniques can be used to determine whether a specific
human CYP enzyme is responsible for the metabolism of specific
drugs and whether specific drugs are capable of altering the
functional activity of a specific human CYP enzyme. Presently,
the most common approach employs microsome preparations. After
an appropriate series of samples have been prepared, a substrate
(eg, a drug known to be metabolized by that enzyme) for the
enzyme is added in a predetermined concentration. Then, the
potential inhibitor is added in varying
concentrations to determine its potency for blocking the enzyme-mediated
biotransformation of the substrate. This approach is analogous
to determining the binding affinity of a drug to a specific
neuroreceptor (see Section 3).
The results of such in vitro studies for the various
SSRIs for 3 important CYP enzymes are shown in Table
8.7.
While the in vitro potency is an important determinant
of the potential for a drug to produce an effect, it is not
the sole determinant. Obviously, the drug must reach a sufficient
concentration at the site of action (SOA) (which is the CYP
enzyme in this instance) to produce sufficient inhibition
of the enzyme to be clinically meaningful. In the case of
a pharmacokinetic interaction, this means the inhibition of
the enzyme must be sufficient to cause a clinically meaningful
change (either an increase or decrease depending on the interaction
in question) in the plasma and tissue concentration of a concomitantly
administered drug which is dependent on that CYP enzyme for
its clearance. There is no simple way to determine whether
the degree of inhibition that will be achieved by two drugs
will be the same or different based solely on their relative
in vitro potency for inhibiting an enzyme. Instead,
the relative concentrations of the drugs under clinically
relevant conditions must be taken into account. Moreover,
it is not the plasma drug concentration that is critical but
rather the concentration at the enzyme.
Mathematical modeling can be done to estimate the degree
of enzyme inhibition that will be produced by a given drug
under clinically relevant conditions based on a knowledge
of 3 factors:
- In vitro inhibition constant of the drug for that
specific enzyme
- Plasma concentration of the drug that occurs under those
conditions
- Plasma:hepatic partition coefficient for that drug under
those conditions
Many readers may not be interested in more details concerning
such modeling work beyond knowing that it exists and what
its implications are. For those who are interested in more
details, see Section 10, Appendix.
The important application of such modeling is that it can
help to determine whether a specific pharmacokinetic interaction
is likely to occur to a clinically meaningful extent when
that drug is coprescribed with a drug that is dependent on
that enzyme for its elimination. This approach can help to
rationally guide research to focus on the most critical pharmacokinetic
studies (ie, those which are likely to reveal an important
interaction). This is a significant advance over the previous
approach where drugs were studied simply because they had
a higher likelihood of being coprescribed together or because
interactions had been described with other drugs in a "class"
such as "antidepressants" which includes vastly
different drugs such as TCAs, SSRIs and monoamine oxidase
inhibitors (MAOIs).
Although such modeling work is an important screening tool
to focus research on highly likely interactions, a definitive
answer as to whether a drug will inhibit a CYP enzyme to a
sufficient extent to produce a clinically meaningful drug-drug
interaction requires formal in vivo pharmacokinetic
interaction studies done under clinically relevant dosing
conditions. In such studies, the degree of enzyme inhibition
is typically measured by changes in the pharmacokinetics of
a drug dependent on a specific CYP enzyme
for its clearance in the absence, and then in the presence,
of the potential inhibitor.
| TABLE 8.7 The Relative
Potency* of Five Different SSRIs and Their Metabolites
for Inhibiting the Functional Integrity of Three CYP Enzymes
1A2, 2D6 and 3A3/4 Based on In vitro Studies Using Human
Hepatic Microsomes |
| Study |
Citalopram / desmethyl-citalopram |
Fluoxetine / Norfluoxetine |
Fluovoxamine |
Paroxetine / M2 |
Sertraline / desmethyl-sertraline |
| |
CYP
1A2 |
|
| Brosen et al1 |
>100/ >100 |
>100/ >100 |
0.2 |
45/NA |
70/NA |
| Rasmussen et al2 |
>100/ >100 |
>100/ >100 |
0.2 |
50 |
>100 |
| von Moltke et al3 |
NA |
4.4/5.9 |
0.24 |
5.5 |
8.8/9.5 |
| |
CYP
2D6 |
|
| Crewe et al4 |
5.1 |
0.60/0.45 |
8.2 |
0.15/0.50 |
0.70/NA |
| Skjelbo et al5 |
19/1.3 |
0.92/0.33 |
3.9 |
0.36/NA |
-- |
| Von Moltke et al6 |
-- |
3.0/3.5 |
16.6 |
2.0/NA |
22.7/16.9 |
| Otton et al7 |
-- |
0.17/0.19 |
-- |
-- |
1.5/NA |
| Otton et al8 |
-- |
0.15/NA |
-- |
0.065/NA |
1.2/NA |
| |
CYP
3A3/4 |
|
| Von Moltke et al9 |
-- |
83.3/11.1 |
10 |
39/NA |
23.8/20.4 |
| Rasmussen et al2 |
>100/ >100 |
60/19 |
40 |
70 |
90 |
* In vitro
potency = Ki = inhibition constant; the smaller
the value, the greater the potency on a molar basis.
Ki values for comparison: quinidine = 626;
ketoconazole = 0.0510 |
| NA = Not applicable |
| References:
147, 2235,
3299, 464,
5256, 6278,
7196, 8194,
9279, 10276 |
In Vivo Studies
The general design for a formal in vivo pharmacokinetic
study is given in Figure 8.1. The essence
of these studies is to determine whether the coadministration
of the potential inhibitor or inducer alters the clearance
of a concomitantly administered drug that is a model substrate
for the enzyme in question. Ideally, such studies use a model
substrate which is a drug that is solely metabolized by one
CYP enzyme and does not itself alter this enzyme's activity
(ie, no autoinduction or autoinhibition) over the clinically
relevant concentration range.
The first step is to give the model substrate to normal
volunteers to determine its rate of biotransformation and
elimination (ie, its clearance) in the absence of the potential
inhibitor or inducer. Two different dosing strategies have
been used with regard to the administration of the model substrate.
In the single dose approach, the clearance of the model
substrate in a volunteer is quantitated after a single dose
in the absence and presence of the potential inhibitor or
inducer. The latter is usually dosed to steady-state prior
to the single-dose rechallenge of the model substrate. In
the multiple dose approach, the model substrate is administered
to the volunteer on a daily basis for a sufficiently long
enough interval to ensure that its steady-state has been achieved.
The potential inhibitor or inducer is then added to the regimen
at a clinically relevant dose and administered sufficiently
long enough to ensure it has also reached steady-state conditions.
The clearance of the model substrate is measured before and
after the addition of the potential inhibitor or inducer.
The multiple dose approach is more rigorous and more expensive,
and more closely mimics clinical practice than does the single
dose approach.
In both approaches, the potential inhibitor or inducer is
administered on a multiple dose basis in an analogous
way to how it would be given in clinical practice. Changes
in the plasma levels and clearance of the model substrate
quantitatively reflect the degree of enzyme inhibition or
induction produced by the potential inhibitor or inducer respectively
under clinically relevant conditions.
| FIGURE 8.1 Design of Pharmacokinetic
Interaction Studies Using SSRIs as an Example |
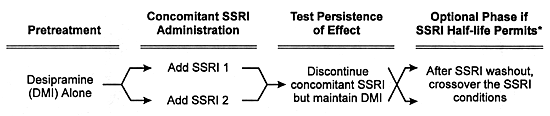 |
|
* Generally impractical with fluoxetine
due to the protracted interval needed for washout of
that SSRI and its effect on CYP enzymes.
|
In the past, in vitro and in vivo studies
have been done in a nonsystematic, nonsequential fashion.
For example, in vivo studies might be done before in
vitro studies or, even though an in vitro or in
vivo study might be positive, the other type of study
might not be done. An in vitro study might show that
a specific drug has an inhibition potency such that one would
expect that the effect could occur under clinically relevant
conditions, but the confirmatory in vivo studies might
not be done. For these reasons, there are gaps in our current
knowledge, but they are in the process of being filled.
The trend now is for these studies to be done using a more
sequential approach. For efficiency, in vitro studies
are done first to screen for potentially meaningful effects
of an investigational drug on a specific CYP enzyme. Those
studies determine the in vitro potency of the investigational
drug for inhibiting or inducing the enzyme. This information,
coupled with a knowledge of what concentration of the investigational
drug is achieved under clinically relevant conditions, can
be used to predict whether this drug is likely to produce
meaningful inhibition or induction of the enzyme under clinically
relevant conditions. Then, in vivo studies are done
to confirm cases where it is likely a pharmacokinetic drug
interaction will occur under such conditions. The in vivo
follow-up studies are done with drugs (ie, model substrates)
that can serve as in vivo probes to quantify the functional
activity of the enzyme before and after the coadministration
of the investigational drug (ie, the potential inhibitor or
inducer). The results of the in vivo studies
can also be used to predict whether other drugs that are substrates
for that specific CYP enzyme will be affected when they are
coadministered with the investigational drug under clinically
relevant conditions.
Obviously, this approach is not restricted to SSRIs but
can be used with any drug suspected of affecting a specific
CYP enzyme. Research with SSRIs has simply been pioneering
work in this area and has documented that several SSRIs are
capable of inhibiting one or more CYP enzyme to a clinically
meaningful extent under clinically relevant conditions.
Beyond this systematic approach to detecting potential pharmacokinetic
drug interactions, spontaneous observation in clinical practice
can lead to case reports. These reports can serve the useful
purpose of guiding formal research, particularly when there
is in vitro data that also supports the probability
of an interaction under clinically relevant conditions. Nonetheless,
case reports are only suggestive and not definitive and clearly
cannot be equated with the results from formal pharmacokinetic
studies due to the limited amount of data and the typical
absence of adequate controls. For example, there is often
no control for compliance in such case reports and the interpretation
is highly dependent on the validity of the first sample obtained,
which is typically done on an outpatient basis.
What Do We Know About the Effects
of Specific SSRIs on Specific CYP Enzymes?
The results from the studies that have been done on 5 major
xenobiotic CYP enzymes (ie, CYP 1A2, 2C9/10, 2C19, 2D6 and
3A3/4) follows and is also summarized in 2 tables. Table
8.8 presents a composite summary based on the available
formal in vitro and in vivo studies. As can
be seen from the table, there is
a variable amount of data with regard to both a specific enzyme
and the effects of a specific SSRI. Data relative to a specific
CYP enzyme may be available for some, but not all, SSRIs.
Data may come solely from either in vitro or in
vivo studies and sometimes data from both types of studies
are available, which is the ideal situation. Occasionally,
only suggestive case reports are available, either alone or
combined with in vitro studies. Again, data based on
case reports, particularly when limited to a small number
of patients, must be interpreted in a cautious manner and
not equated with results from formal, rigorous studies. Table
8.7 summarizes the results of the in vitro studies
that have been done with the SSRIs relative to three specific
enzymes: CYP 1A2, 2D6 and 3A3/4. Tables
8.9 and 8.11
summarize the in vivo studies.
| TABLE
8.8 Effects of Specific SSRIs on Specific CYP Enzymes
at Their Respective, Usually Effective, Antidepressant
Dose* |
| Enzyme |
Citalopram |
Fluoxetine |
Fluvoxamine |
Paroxetine |
Sertraline |
| CYP 1A2 |
Unlikely1 |
Unlikely1 |
Substantial1,2 |
Unlikely1
|
Unlikely1 |
| CYP 2C9/10 |
? |
?3 |
? |
NCS2 |
NCS2 |
| CYP 2C19 |
? |
Moderate2 |
Substantial2 |
? |
NCS2 |
| CYP 2D6 |
Mild1 |
Substantial1,2 |
NCS1,2 |
Substantial1,2 |
Mild1,2 |
| CYP 3A3/4 |
? |
Mild1,2 |
Moderate1,2 |
Unlikely1,2 |
Unlikely1,2,4 |
? = absence of or
contradictory in vitro or in vivo data available for this
SSRI
Unlikely = based on in vitro studies, unlikely to have
a clinically meaningful effect
NCS = not clinically significant in most situations =
< 20% change
| 1Change in
the area under the curve (AUC) of the plasma level-time
curve of a substrate (ie, concomitantly administered
drug) dependent on that CYP enzyme for its clearance. |
Mild = 20% to 50% change
Moderate = 50% to 150% change
Substantial = > 150% change |
|
* Table is a summary of the results
presented in Tables 8.7, 8.9,
8.11, and 10.1,
and data reviewed in the text. Hence, it is based on
effects observed or predicted based on the concentration
of these drugs that would be usually produced by their
usually effective, antidepressant dose. Since the inhibition
of these enzymes is concentration-dependent, the magnitude
of the effect will be higher on average at higher doses
particularly for SSRIs with nonlinear pharmacokinetics
(see Table 6.2).
|
- Predicted based on in vitro inhibitory rate constants
and on knowledge about clinically relevant plasma
drug levels (for details about predictive modelling,
see Section 10, Appendix).
- Based on the following in vivo data: A large number
of well documented case reports principally involving
theophylline for fluvoxamines effect on CYP 1A2 and
formal in vivo studies is summarized in Table
8.9 for effects on CYP 2D6 and Table
8.11 for effects on CYP 2C9/10, 2C19, and 3A3/4.
Relative to CYP 3A3/4, data with paroxetine and sertraline
are limited to carbamezapine rather than a more model
substrate such as alprazolam.
- Studies with tolbutamide and warfarin suggest no
effects, but 23 cases with adequate data with phenytoin
suggest a substantial effect of fluoxetine on CYP
2C9/10. The reasons for the discrepancy in these results
are not clear.
- The designation, unlikely, is further supported
by the linear pharmacokinetics of sertraline over
its full dosing range (ie, 50 to 200 mg/day) since
sertraline is demethylated by CYP 3A3/4 (see Section
6).
|
The data regarding the differential effects of SSRIs on
CYP 1A2 come principally from 2 in vitro studies (Table
8.7). Brosen et al and Rasmussen et al have shown that
fluvoxamine has an inhibitory rate constant for this enzyme
such that it is likely to cause clinically meaningful inhibition
under antidepressant treatment conditions (Table
8.7). Consistent with these in vitro findings,
there is evidence that fluvoxamine inhibits the clearance
of drugs dependent on this enzyme for biotransformation prior
to elimination. While the rate-limiting step for the elimination
of all TCAs is ring hydroxylation mediated by CYP 2D6,14,15,25,38,40-42,45,46,48,67,109,161,171,192,195,205,262
the tertiary amine TCAs are demethylated by several CYP enzymes
including CYP 1A2, 3A3/4 and possibly 2C19. 19,38,41,42,46,58,118,161,168,192,202,247,256,257,262
Concomitant administration of fluvoxamine inhibits such
demethylation and produces higher than usual concentrations
of the parent drug (ie, tertiary amine TCAs such as clomipramine)
relative to their demethylated metabolite (ie, a secondary
amine TCA such as desmethylclomipramine) (see Section
3).18,27,28,118
Fluvoxamine can also produce clinically significant elevations
of theophylline and clozapine, drugs dependent on CYP 1A2
for their metabolism.137,260,270,274
| TABLE 8.9 In Vivo
Studies of Effects of Different SSRIs on CYP 2D6 Function |
| # |
Study |
Reference |
Drug |
Dose |
Substrate |
Result |
| 1 |
Gram et
al, 1993 |
110 |
Citalopram |
40 mg/d |
IMI/DMI |
 47%
47% |
| 2 |
Bergstrom
et al, 1992 |
24 |
Fluoxetine |
60 mg/d
(7 days) |
DMI |
640% |
| 3 |
Preskorn
et al, 1994 |
219 |
Fluoxetine |
20 mg/d
(3 wks) |
DMI |
380% |
| 4 |
Spina et
al, 1993 |
263 |
Fluovoxamine |
100 mg/d |
IMI/DMI |
14% |
| 5 |
Albers et
al, 1995 |
4 |
Paroxetine |
30 mg/d |
IMI/DMI |
327% |
| 6 |
Alderman
et al, 1996 |
5 |
Paroxetine |
20 mg/d |
DMI |
421% |
| 7 |
Brosen et
al, 1993 |
41 |
Paroxetine |
20 mg/d |
DMI |
364% |
| 8 |
Jann et al,
1996 |
136 |
Sertraline |
50 mg/d |
IMI |
0% |
| 9 |
Preskorn
et al, 1994 |
219 |
Sertraline |
50 mg/d |
DMI |
23% |
| 10 |
Alderman
et al, 1996 |
5 |
Sertraline |
50 mg/d |
DMI |
37% |
| 11 |
Sproule et
al, 1995 |
264 |
Sertraline |
108 mg/d |
Dextromethorpan |
5% |
| 12 |
Zussman
et al, 1994 |
298 |
Sertraline |
150 mg/d |
DMI |
54% |
| 13 |
Kurtz et al, 1994 |
153 |
Sertraline |
150 mg/d |
DMI |
70% |
| TABLE 8.10 Relative
Potency of the Enantiomers of Fluoxetine and Norfluoxetine
for Inhibiting the CYP enzyme 2D6 |
| Enantiomer |
Kinetic inhibitory constant
Ki (µm)1 |
| S-fluoxetine |
0.22 |
| R-fluoxetine |
1.38 |
| S-norfluoxetine |
0.31 |
| R-norfluoxetine |
1.48 |
|
S/R ratio for fluoxetine and norfluoxetine
= 2.22
|
|
S/R ratio = ratio of plasma levels
of the 2 enantiomer under steady-state conditions when
the racemic mixture is being taken.
|
|
Data from references: 1267,
2272
|
Based on the results from the Brosen et al and Rasmussen
et al in vitro studies, the other
SSRIs would not be expected to inhibit CYP 1A2 to any meaningful
extent under clinically relevant conditions (Table
8.7). While there are no formal in vivo studies,
there is indirect in vivo evidence to support this
conclusion. Although concomitant administration of fluvoxamine
produces a substantial increase in warfarin plasma levels
(+ 65%),22 this effect
does not occur with fluoxetine, paroxetine or sertraline.16,245,287
Warfarin is a racemic mixture. S-warfarin is metabolized
by CYP 2C9/10 and is the active enantiomer in
terms of anticoagulant effect.149,162,238
The inactive R-warfarin enantiomer is metabolized by
CYP 1A2 but can inhibit CYP 2C9/10 and thus produce an accumulation
of the active S-warfarin enantiomer. The fact that
fluvoxamine produces a buildup of warfarin plasma levels is
compatible with the inhibition of CYP 1A2 and a resultant
buildup of R-warfarin plasma levels which in turn inhibit
CYP 2C9/10, resulting in a buildup of S-warfarin plasma
levels and hence, an increase in anticoagulant effect. The
fact that this scenario does not occur with sertraline, fluoxetine
and paroxetine is compatible with an absence of effect of
these 3 SSRIs on both CYP 1A2 and 2C9/10.
There have been no in vitro studies of the different
effects of SSRIs on CYP 2C9/10. However, in vivo studies
have been done with both tolbutamide and warfarin, which are
substrates for this enzyme (Table 8.11).
Fluoxetine and sertraline did not produce a clinically significant
decrease in the clearance of tolbutamide or warfarin, and
paroxetine did not affect warfarin.16,159,245,273,287
Despite these results with fluoxetine, the U.S. Food and Drug
Administration has compiled 163 cases in which the concomitant
use of fluoxetine elevated phenytoin plasma levels. In 23
cases with adequate data, the average increase was 161%, occurring
on average 2 weeks after fluoxetine was started, and was associated
with development of clinical manifestations of toxicity including
ataxia, somnolence and nystagmus.71,134,249,293
These cases have led to a revision of the product labelling
for fluoxetine to advise about this interaction. Since phenytoin
is metabolized by CYP 2C9/10, this interaction suggests that
fluoxetine inhibits this CYP enzyme. The reason for the apparent
discrepancy between this effect of fluoxetine on phenytoin
clearance and the absence of an effect
of fluoxetine on the clearance of tolbutamide and warfarin
is not known.
No in vivo studies have been done with SSRIs using
an ideal model substrate for this CYP enzyme. However, studies
with 3 SSRIs (ie, fluoxetine, fluvoxamine and sertraline)
have been performed with diazepam which is principally dependent
on CYP 2C19 for its metabolisms at least at low concentrations
(Table 8.11). Demethylation of diazepam
to desmethyldiazepam is the major route of elimination and
is dependent on CYP 2C19 at the concentrations achieved on
conventional low doses.9,26,56,132,286
At higher concentrations, this demethylation can also be mediated
by CYP 3A3/4.10,294
Hydroxylation of diazepam to temazepam is typically a minor
pathway and is mediated by CYP 3A3/4.26,119
Desmethyldiazepam undergoes C3-hydroxylation which may be
mediated by a single, rate-limiting enzyme, CYP 2C19.26,119
In contrast, C3-hydroxylation of diazepam is minimal at low
drug concentrations, increases sigmoidally with increasing
concentrations, and may be mediated entirely by CYP 3A3/4.119
Diazepam thus does not fulfill the criteria of a model substrate
due to its multiple pathways mediated by more than one CYP
enzyme. Nonetheless, the in vivo studies that have
explored the potential interaction between these three SSRIs
and diazepam does provide useful information on whether they
inhibit either CYP 2C19 or 3A3/4. Since the metabolism of
diazepam at low substrate concentrations appears to be principally
mediated by CYP 2C19, and since the in vivo studies
that follow were done with low doses of diazepam and hence,
at low substrate concentrations, their findings suggest the
delay in diazepam clearance produced by some SSRIs is due
to an effect on CYP 2C19.
Fluvoxamine (average dose = 112 mg/day) produced on average
a 280% and 41% increase in the area under curve (AUC) for
diazepam and desmethyldiazepam respectively. The effect on
diazepam represented a 65% reduction in its apparent clearance
from 0.40 to 0.14 ml/min/kg and a marked prolongation in diazepam
half-life from 51 to 118 hours (p < 0.01).203
Fluoxetine, using a loading dose strategy (60 mg/d ×
8 days) that would produce combined levels of fluoxetine and
norfluoxetine approximately 20% to 30% lower than those that
will occur at steady-state on 20 mg/day, produced a 40% increase
in the AUC of diazepam.160
Another study using only 30 mg/d × 7 days did not produce
an increase in diazepam AUC.159
This finding is consistent with the concentration-dependent
nature of the effect since this loading dose strategy (30
mg/d × 7 days) would produce less than half the levels
that would be expected under steady-state conditions on 20
mg/day. In contrast to fluvoxamine, fluoxetine (60 mg/d ×
8 days) reduced the levels of metabolically derived desmethyldiazepam;
which raises the possibility of a different MOA (ie, an effect
on a different CYP enzyme such a CYP 3A3/4 rather than CYP
2C19). Further work is needed with fluoxetine and a more ideal
substrate for CYP 2C19 to determine to what extent its effects
on diazepam are due to an effect on CYP 2C19 versus CYP 3A3/4.
Based on the study by Gardner and colleagues, sertraline under
steady-state conditions on its usually effective, minimum
dose of 50 mg/day will be expected to produce an appreciably
smaller effect (ie, approximately 8% increase in AUC) than
either fluvoxamine or fluoxetine.99
Thus, these studies with these 3 SSRIs suggests that CYP 2C19
is inhibited potently by fluvoxamine, possibly weakly by fluoxetine,
and trivially inhibited by sertraline at each drugs' usually
effective, minimum, antidepressant dose. No comment can be
made about the potential impact of citalopram or
paroxetine on CYP 2C19 since appropriate studies have not
been done.
A large number of drugs from different therapeutic classes
are dependent on 2D6 for their metabolism including TCAs,
codeine, several neuroleptics, B-blockers, and Type
IC antiarrhythmics (Table 7.9).
Of all the CYP enzymes, 2D6 has been studied most thoroughly
with respect to the different effects of SSRIs on its functional
activity (Table 8.7, Table
8.9 and Figure 8.2). Based on most
in vitro studies (Table 8.7),
paroxetine is 2- to 4-times more potent than fluoxetine, and
5 to 20 times more potent than sertraline. Fluvoxamine and
citalopram are also substantially less potent than paroxetine
and fluoxetine with respect to the in vitro inhibition
of 2D6.
Recall that fluoxetine is marketed as the racemate (Section
2). The in vitro data shown in Table
8.7 for the inhibition of CYP 2D6 is for the racemate.
The potency of the two enantiomers of fluoxetine and norfluoxetine
is shown in Table 8.10. The relative
potency of these enantiomers for other CYP enzymes has not
been established.
All of SSRIs, with the apparent exception of fluvoxamine,
have "active" metabolites relative to the inhibition
of this enzyme in vitro with a potency similar to that
of the parent compound (Table 8.7).
However, only norfluoxetine reaches sufficient concentrations
at each drug's usually effective, minimum concentration under
routine clinical conditions to contribute in a clinically
meaningful way to the inhibition of this enzyme. The contribution
of norfluoxetine takes on special significance due to its
extended half-life (ie, 1 to 2 weeks), which means that the
risk of a drug-drug interaction mediated by this active metabolite
persists for weeks
after fluoxetine has been discontinued.
| FIGURE 8.2 Differential In
Vivo Effects of Five Different SSRIs on CYP 2D6 Function |
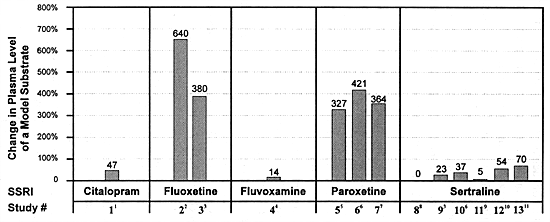 |
| References: 1110,
224, 3219,
4263, 54,
65, 741,
8136, 9264,
10298, 11153 |
Desipramine has generally served as the
model substrate for the in vivo studies of SSRIs on
CYP 2D6 function (Table 8.9 and Figure
8.2). In these studies, the effect of each SSRI on the
clearance of desipramine has been studied primarily at their
usually effective dose: fluoxetine and paroxetine (20 mg/day)
and sertraline (50 mg/day) (see Section
5). Since a fixed-dose study has not been published for
citalopram or for fluvoxamine, their usually effective, minimum
doses have not been as rigorously established (see Section
5). Nevertheless, they have been tested for effects on
CYP 2D6 at doses of 40 mg/day for citalopram and 100 mg/day
for fluvoxamine.
The results are quite clear about the relative impact of
each SSRI on the in vivo clearance of desipramine:
fluoxetine produced a 380% to 640% increase in desipramine
levels; paroxetine, a 327% to 421% increase; citalopram, a
47% increase; sertraline, a 0% to 37% increase; and fluvoxamine,
a 14% increase (Table 8.9, Figure
8.2). Thus, only 2 out of the 5 SSRIs marketed worldwide
produce meaningful inhibition of this enzyme at their usually
effective dose.
Only in the broadest sense can one state that all the SSRIs
inhibit CYP 2D6. It is misleading to make such a claim without
acknowledging the substantial differences among these drugs.
To further put the differences among the SSRIs in perspective,
the above results for fluoxetine somewhat underestimate its
full effect at steady-state at 20 mg/day. That is because
steady-state levels of fluoxetine and norfluoxetine that would
be expected at 20 mg/day were not achieved in either study
due to the extended half-life of norfluoxetine. That is true
even for the Bergstrom et al study which used a loading dose
strategy of 60 mg/day for 7 days.
Only paroxetine and sertraline have been formally studied
at doses above their usually effective dose. The study by
Albers et al used a dose of 30 mg/day;
however, that dose was administered for only 4 days prior
to the administration of the test dose of imipramine.4
Consistent with that short treatment phase, the plasma levels
of paroxetine in this study (10-30 mg/ml) were similar to
the 2 studies which gave 20 mg per day for at least 10 days.5,41
Thus, the study by Albers et al was more of replication of
those 2 earlier studies than a test of a higher dose.
Sertraline has been the one SSRI most extensively tested
at doses above its usually effective antidepressant dose.
Sproule et al found a 5% prolongation in the clearance of
dextromethophan following treatment with sertraline 100 mg/day
for 21 days.264
Two separate studies examined the effect of sertraline 150
mg/day administered for a sufficient duration to achieve steady-state
and found a 54% to 70% increase in desipramine plasma levels.153,298
Thus sertraline at its usually effective dose of 50 mg/day
produces a 0% to 37% increase in the plasma levels of a model
substrate such as desipramine which is principally dependent
on CYP 2D6 for its clearance, a 5% prolongation in clearance
at 108 mg/day, and a 54% to 70% increase in plasma levels
at 150 mg/day. The magnitude of this effect is substantially
less at all of these doses than the effect of fluoxetine and
paroxetine at their usually effective minimum dose.
While citalopram and fluvoxamine have been less extensively
studied than sertraline, their effects on
CYP 2D6 are also substantially less than fluoxetine and paroxetine
(Table 8.9, Figure
8.2).
| FIGURE 8.3 Trough Concentrations
of Desipramine in Plasma Correlated With Concentrations
of Sertraline Plus Desmethylsertraline in the Sertraline
Treatment Group and Fluoxetine Plus Norfluoxetine
in the Fluoxetine Treatment Group |
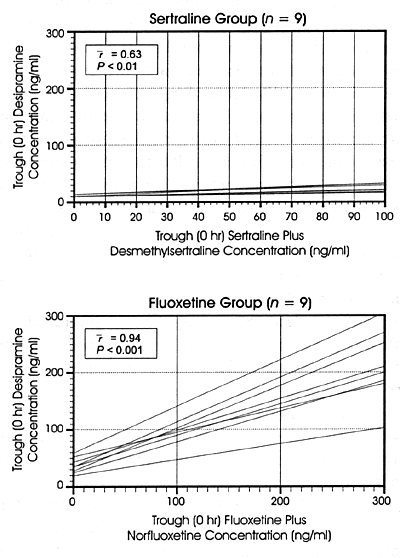 |
| Reference: 219 |
|
| FIGURE 8.4 Trough Concentrations
of Desipramine in Plasma Correlated With Concentrations
of Sertraline Plus Desmethylsertraline in the Sertraline
Treatment Group and Paroxetine in the Paroxetine
Treatment Group |
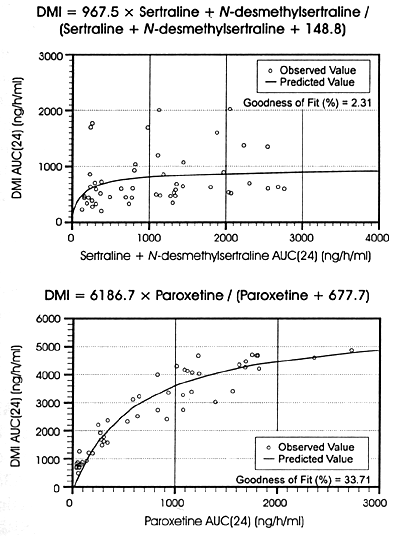 |
|
Michaelis-Menten Modeling of
desipramine AUC(24) to sertraline + N-desmethylsertraline
AUC(24) and Paroxetine AUC(24).
|
| AUC = Area under curve. |
| Reference: 5 |
|
Figures 8.3 and 8.4
demonstrate the concentration-dependent nature of the effects
of fluoxetine, paroxetine and sertraline on CYP 2D6 as reflected
in the changes in plasma levels of the CYP 2D6 substrate,
desipramine. These figures are from the formal in vivo
pharmacokinetic studies that compared the effects of fluoxetine
20 mg/day to sertraline 50 mg/day in one study219
and the effects of paroxetine 20 mg/day to sertraline
50 mg/day in a second study.5
On the X-axis of the graphs is the concentration of each respective
SSRI and on the Y-axis is the plasma concentration of desipramine.
There is a robust relationship between increasing plasma levels
of fluoxetine plus norfluoxetine and increasing levels of
desipramine in Figure 8.3. The same
is true for paroxetine and desipramine plasma levels in Figure
8.4. In both figures, the relationship between increasing
plasma levels of sertraline and elevations of desipramine
plasma levels is considerably more modest. These results are
fully consistent with the differences in the in vitro potency
of these 3 SSRIs for the inhibition of CYP 2D6 and the differences
in concentrations of 3 SSRIs at this enzyme at each drug's
usually effective, minimum dose. Table
10.1 provides further illustration of the concentration-dependent
nature of the inhibition of CYP enzymes.
To put these results in perspective, fluoxetine and paroxetine
appear to produce approximately 85% inhibition of CYP 2D6
activity versus 15% inhibition or less with citalopram, fluvoxamine
or sertraline (Table 8.4).
With the exception of citalopram, all of the SSRIs and their
primary metabolites have been studied in terms of their in
vitro effects on the metabolism of alprazolam, which is
a substrate for the CYP 3A3/4 enzyme (Table
8.7). Fluvoxamine and norfluoxetine were the most potent
in vitro inhibitors of the SSRIs. Sertraline and paroxetine
were intermediate. Fluoxetine, the parent drug, was the weakest.
To keep these results in perspective, the in vitro
potency of both fluvoxamine and norfluoxetine are considerably
less potent than the antifungal agent, ketoconazole.278
This antifungal agent can produce clinically serious elevations
of terfenadine due to its inhibition of
CYP 3A3/4.29,126,127,279,297
Based on the results of in vitro studies, coadministration
of fluvoxamine and fluoxetine (due to norfluoxetine) would
be predicted to cause more in vivo inhibition of CYP
3A3/4-dependent drug metabolism than would either paroxetine
or sertraline due to the difference in plasma concentrations
of these different agents at comparable
antidepressant doses of each agent (Table
8.4 and Section 10, Appendix)
but substantially less than ketoconazole.
Unfortunately, this prediction has only been partially tested
by formal in vivo studies. The in vivo effects
of fluoxetine and fluvoxamine on the clearance of alprazolam,
a model substrate for CYP 3A3/4, has been tested in vivo,
but similar studies have not been done with citalopram, paroxetine
or sertraline (Table 8.11). Fluvoxamine
(100 mg/day for 10 days) produced a doubling of alprazolam
plasma levels with a 55% decrease in its clearance.87
This finding is compatible with the in vitro data and
the known plasma levels of fluvoxamine that would be expected
at this dose.
In the first study with fluoxetine, a 33% increase in alprazolam
plasma levels occurred after only 4 days of coadministration
of fluoxetine 60 mg/day (ie, a loading dose strategy).157
In the second study, the loading dose strategy for fluoxetine
was 40 mg/day for 10 days which produced a combined plasma
level of fluoxetine and norfluoxetine of 160 ng/ml, which
is approximately 25% less than the steady-state levels of
200 ng/ml that should occur on 20 mg/day.112
In this study, fluoxetine produced a 25% decrease in the clearance
of alprazolam. Based on the results of these 2 studies, fluoxetine,
at steady-state on 20 mg/day, will be expected to produce
a 30% to 40% increase in alprazolam plasma levels. As with
the inhibition of CYP 2D6, the effect of norfluoxetine on
CYP 3A3/4 can last for an extended interval after fluoxetine
discontinuation. In the second study, the increase in alprazolam
plasma levels persisted for more than 2
weeks after norfluoxetine was discontinued.112
There have been some other studies relative to CYP 3A3/4
substrates which deserve some comment (Table
8.11). Carbamazepine is in part metabolized by CYP 3A3/4.143,172,207
However, carbamazepine is not a model substrate due to the
fact that it induces its own metabolism as well as the metabolism
of other drugs. Thus, interaction studies with carbamazepine
are not easily interpreted. With this caveat in mind, it is
noteworthy that fluoxetine-induced slowing of carbamazepine
metabolism has been reported by some but not all investigators.101,102,113,201,261
There is also a case report describing cardiac abnormalities
occurring 30 days after fluoxetine was added to a regimen
containing terfenadine and resolved when terfenadine was stopped.268
This case, coupled with the in vitro and in vivo
data discussed above, suggests the need for a formal study
given the widespread use of fluoxetine and terfenadine, coupled
with the potential seriousness of a significant interaction.
The available data suggest that fluoxetine at 20 mg/day is
unlikely to produce sufficient inhibition of CYP 3A3/4 to
produce a clinically significant interaction with terfenadine.
| TABLE 8.11 Comparison
of the In Vivo Effects of Different SSRIs on Specific
CYP Enzyme substrates* |
| SSRI |
Changes in Plasma
Levels |
| CYP
2C9/10 |
CYP 2C19 |
CYP
3A3/4 |
| TBA |
PHT |
DZ |
APZ |
CBZ |
| Fluoxetine |
4%1 |
161%2 |
50%3 |
33%4
26%5 |
27%6
0-63%7 |
| Fluvoxamine |
NA |
NA |
300%8 |
100%9 |
30-70%10 |
| Paroxetine |
0%11 |
NA |
NA |
NA |
0%12 |
| Sertraline |
 5%13
5%13 |
0%14 |
3%15 |
NA |
0%16 |
|
* CYP 2C9/10: TBA = tolbutamide, PHT
= phenytoin
CYP 2C19: DZ = diazepam
CYP 3A3/4: APZ = alprazolam, CBZ = carbamazepine
|
- Formal in vivo study, 30 mg/d × 8 days, an inadequate
loading dose strategy.159
- Large number of case reports with doses generally
20 mg/d for weeks.71,134,249,293
- Formal in vivo study, 60 mg/d × 8 days.160
- Formal in vivo study, 60 mg/d × 4 days.157
- Formal in vivo study, 40 mg/d × 7 days.112
- Formal in vivo study, 20 mg/d × 7 days.113
- Large number of case reports with doses generally
approximately 20 mg/d at steady-state.101,201,261
- Formal in vivo study, 112 mg/d × 16 days.203
- Formal in vivo study, 100 m/d × 10 days.87
- Large number of case reports with doses of 100-300
mg/d.35,96
- Formal in vivo study, 30 mg/d × 10 days. While
there was a non-significant increase in warfarin AUC,
5 out of 27 experienced increased bleeding.16
- Controlled-case series, 30 mg/d × 10 days.8
- Formal in vivo study, 200 mg/d × 16 days caused
an 18% increase which would be approximately 5% at
its usually effective dose of 50 mg/d.273
- Formal in vivo study, 200 mg/d × 16 days. No significant
change.65
- Formal in vivo study, 200 mg/d × 16 days caused
a 13% increase which would be approximately 3% at
its usually effective dose of 50 mg/d.99
- Formal in vivo study, 200 mg/d × 16 days caused
a nonsignificant decrease.74
|
There are also case reports of 30% to 70% increases in carbamazepine
levels when 100 to 300 mg/day of fluvoxamine is coadministered.35,96
In a formal study, sertraline at a dose of 200 mg/day (which
is four-times its usually effective, minimum dose) did not
alter carbamazepine levels.74
In a controlled-case series as opposed to a formal pharmacokinetic
study, paroxetine did
not alter carbamazepine levels.8
Taken as a whole, these studies indicate that CYP 3A3/4
inhibition produced by SSRIs at their usually effective dose
ranges from moderate to not detectable as follows: fluvoxamine
(moderate) > fluoxetine (mild) > paroxetine and sertraline
(not detectable) (Table 8.7). There
are a few caveats to this statement. First,
citalopram is not mentioned because it has not been studied
adequately. Second, the effects on these enzymes is concentration
dependent so that higher doses could produce greater effect.
This is particularly important for SSRIs, which have nonlinear
pharmacokinetics (see Section 7).
For example, fluoxetine produces mild inhibition of CYP 3A3/4
at 20 mg/day, but will be expected to increase disproportionately
with dose increases. The mild inhibition (20% to 50% change)
will not be expected to produce a clinically meaningful drug-drug
interaction in most instances, but the likelihood and severity
of a clinically meaningful interaction increases as the magnitude
of the change in clearance due to the degree of enzyme inhibition
increases (see Appendix).
To put these results in perspective, it is important to
recognize the substantial difference between the inhibition
produced by fluvoxamine and fluoxetine and the effects of
drugs such as ketoconazole. That has unfortunately not been
done in several recent review articles.301,302
These papers have listed several SSRIs as inhibitors of CYP
3A3/4 along with drugs such as ketoconazole without acknowledging
the difference in the magnitude of their effects. The profound
inhibition of CYP 3A3/4 enzyme produced by ketoconazole and
itraconazole is responsible for the potentially fatal interaction
of these drugs with terfenadine. While fluvoxamine is the
most potent SSRI in vitro with regard to
CYP 3A3/4 inhibition, it is three orders of magnitude less
potent than ketoconazole. In vivo studies
have confirmed that there is a 20- to 100-fold difference
between the effects of fluvoxamine and fluoxetine on the CYP
3A3/4 mediated biotransformation of model substrates such
as triazolobenzodiazepines and the effects of antifungal agents
such as ketoconazole and itraconazole under clinically relevant
dosing conditions (Figure 8.5). Failure
to recognize these substantial differences can cause significant
confusion about the relative risk of experiencing a serious
adverse drug-drug interaction due to CYP 3A3/4 inhibition.
| FIGURE 8.5 Relative In Vivo
Effects of CYP 3A3/4 Inhibitors on Triazolobenzodiazepines
(TBZ) |
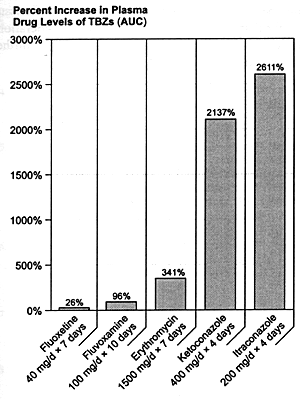 |
| AUC = Area under curve. |
| References: 303-306 |
Table 8.11 summarizes the in vivo
data which is available for the 4 most extensively
studied SSRIs in terms of their effects on CYP enzymes other
than CYP 2D6. The results for CYP 2D6 are summarized in Table
8.9 and Figure 8.2. Since the inhibition
of these enzymes is concentration-dependent, it is important
to ensure an adequate trial of the usually effective, minimum
dose, particularly for SSRIs such as fluvoxamine and fluoxetine
which inhibit more than one CYP enzyme. The data are clear
that the effect of the SSRIs on specific CYP enzymes is an
SSRI-specific issue rather than a class issue and involves
considerations beyond the in vitro inhibition constant
(Ki) as further discussed in Section
10, Appendix.
|



